


抗体药物是一种重要的生物制剂,用来医治多种疾病,如肿瘤、自身免疫性疾病等。抗体药物的生产的全部过程需要经过多个步骤,包括细胞株选择与构建、培养基与生物反应器的选择、细胞培养、抗体表达和纯化、抗体药物制剂
自1999年国内第一款抗体药物“抗人T细胞CD3鼠单抗”国内上市以来,后面上市几十款抗体主要以进口为主,国产抗体在近五年迎来爆发期,创造新兴事物的能力明显提高,但抗体药物产业链上游主要原材料、设备等长期以来高度依赖进口,国内产商寻找技术突破,在分析设备、耗材、培养设备、培养基等多领域实现国产替代,促进抗体药物研发创新。
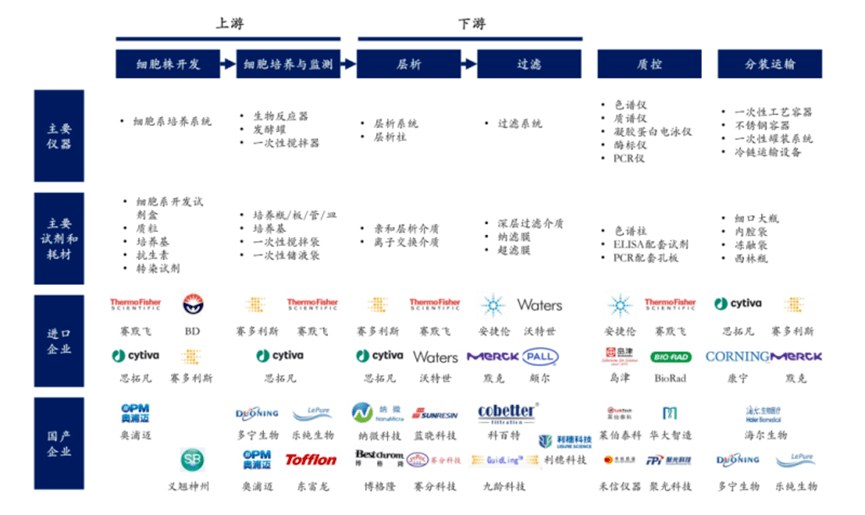
细胞培养是抗体药物生产的第一步,主要是通过培养细胞系来产生目标抗体。哺乳动物细胞是临床抗体药物产品使用的主要表达系统。对于治疗性抗体而言,为满足其生物活性,有必要进行正确的折叠和翻译后修饰,因此用来生产治疗性抗体的宿主细胞往往是哺乳动物细胞,最重要的包含:Sp2/0骨髓瘤细胞、NS0小鼠骨髓瘤细胞、HEK293人胚胎肾细胞和中国仓鼠卵巢细胞(CHO),其中以CHO细胞用途最为广泛。
(5)CHO细胞是成纤维细胞,几乎不分泌内源性蛋白, 因此对目标重组抗体的分离纯化工作十分有利。
抗体药开发是一个很复杂的过程,而构建适用于工业生产的高表达的细胞株是最关键的步骤之一,它直接影响了生产的效率和目标产品质量。因此,构建及优化稳定的表达不同外源基因的重组工程细胞株的实验研究至关重要。目前,治疗性重组蛋白在许多诊断和治疗中应用广泛。与合成药物不同,蛋白药物多来源于原核或真核表达系统。如大肠杆菌、哺乳或非哺乳动物细胞、昆虫细胞等细胞系表达系统,以及近几年热门研究的无细胞表达系统。
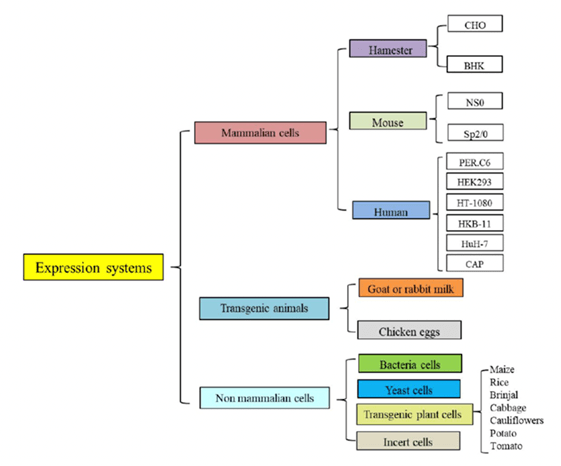
成功的构建工程化细胞株,即从分离高产的哺乳动物细胞株至得到未来用于临床和工程化生产的高产细胞株库,是一个步骤繁琐且漫长的过程,需要做大量的工作才能得到较高产的细胞株。以传统的筛选流程来说,从转染开始至minipool筛选,再到最后的单克隆筛选往往就要花费3-6个月的时间,而后还要进行稳定性传代实验以研究其稳定性,在得到高产单克隆后还要在上游细胞培养工艺开发阶段进行详细的优化与表征,综合起来其研发周期相对于同类药物来说显著延长,依各公司研发实力不同,其细胞株筛选至细胞培养工艺开发结束普遍需要6-12个月,甚至更长。而细胞株的构建在整个抗体产量的流程中占有较小的成本,但是在提高大批量表达的产量时却占有十分重要的地位。目前大部分的抗体药物大都是由CHO细胞进行表达的。CHO细胞可以有效的进行正确的折叠、组装和进行类似于人抗体的翻译后修饰等能力,是生产重组蛋白类药物主要的宿主细胞。
细胞株开发的过程中,通常要在一定的培养工艺平台基础上进行克隆筛选。中试生产申报药品临床试验( investigational new drug,IND) 时,一般会选定2~3个来自不同系列的克隆,其中1~2个克隆作为备选。
而培养工艺对最终生物制品的产量、质量和安全有巨大影响,其中以细胞培养基的选择最重要,配制适合细胞生长和表达的培养基,包括基础培养基和补充物,常用的培养基包括DMEM、RPMI 1640 等。在筛选培养基和补料培养基时,初期流加培养时基础培养基和同品牌的补料培养基应该配对进行筛选。初步筛选之后,将基础培养基和补料培养基进行分类,如促进生长类型、维持活率类型、促进表达类型等。将培养基进行混合优化时,尽量将不一样的培养进行组合,并且应用实验设计( design of experiment,DOE) 优化。
当前,以Amgen、Genentech公司为代表的生物制药巨头所使用的细胞株的蛋白表达量已达到5~10g/L,Pfizer、MedImmune 公司的个别细胞株甚至超过了10g/L。因此,细胞培养的生物反应器也从过去的大型化(20 kL)向小型化(1kL)、连续性、一次性转变。传统的不锈钢反应器在使用前有必要进行彻底的清洁灭菌,而且有着更大的污染的风险,而一次性反应器可以大幅度节省准备的时间。带有搅拌器的袋式生物反应器,如 Hyclone 公司的 SUB、Sartorius公司的 BIOSTAT 和 Xcellerex 公司的 XDR-DSTB,正慢慢的变多地被运用于治疗性抗体的生产。
以典型的单抗药物的商业化生产流程为例,上游工艺(细胞培养)一般都会采用以下步骤:细胞复苏与传代→扩增培养→细胞大规模培养→细胞分离(可属于下游工艺)。
将种子放置在恒温的水浴锅中,一般温度控制在如35℃左右,预热如30min,将工作细胞库细胞进行解冻复苏,根据工艺要求控制解冻时间,然后将解冻细胞加入至培养基中,离心分离后转入摇瓶中(如250 mL),进行摇瓶传代。注意控制CO 2 浓度、培养温度、培养时间等因素,同时也要根据工艺的要求控制接种的密度,控制细胞活率等要求。将上述的复苏种子细胞按照一定的密度稀释至新摇瓶中(如500 mL/2000mL),控制温度、CO 2 浓度、溶氧浓度、培养温度、培养时间、摇床摇摆速度、pH等因素,逐渐扩增细胞数量至满足一级种子罐的接种要求。
将摇瓶种子细胞按照一定的密度接种到一级种子罐内(如20 L),控制温度、CO 2 浓度、溶氧浓度、培养温度、培养时间、搅拌速度、pH、培养基等因素,逐渐扩增至细胞数量满足二级种子罐的要求。
将一级种子按照一定的密度接种到二级种子罐内(如50 L),控制温度、CO 2 浓度、溶氧浓度、培养温度、培养时间、搅拌速度、pH、培养基等因素,逐渐扩增至细胞数量满足三级种子罐的接种要求。
将二级种子按照一定的密度接种到三级种子罐内(如100 L),控制温度、CO 2 浓度、溶氧浓度、培养温度、培养时间、搅拌速度、pH、培养基等因素,逐渐扩增至细胞数量满足细胞发酵培养的接种要求。
种子扩增后要进入细胞大规模培养阶段,注意控制温度、CO 2 浓度、溶氧浓度、培养温度、培养时间、搅拌速度、pH、培养基等因素。细胞培养结束后,可采用离心分离或澄清过滤等办法来进行抗体蛋白的分离纯化。
质量源于设计(QbD) 核心理念在于“质量是通过科学合理的设计得以实现的,而不是仅仅依靠最终检验出来的”。自2013年起,美国 FDA 对于新的药品批准申请,监管部门均要求采用QbD方法。近年来,QbD的理念已慢慢的变成为国内企业大多数药品生产和研发企业共识,在单抗制品的细胞培养工艺开发中应用的报道慢慢的变多,基于QbD理念的药品开发基本流程,ICH Q8中提到的质量源于设计QbD的方法如下图所示:
基于QbD理念的细胞培养工艺开发:首先要界定目标产品的质量概况(QTPP),即以预先设定的目标产品质量概括(QTPP)为研发的起点,在了解关键物质属性(CMA)的基础上,通过试验设计(DOE),理解产品的关键质量属性(CQA),确立关键工艺参数(CPP),在原料特性、工艺条件、环境等多个影响因素下,建立能满足产品性能的且工艺稳健的设计空间(DS),并根据设计空间,建立质量风险管理,确立质量控制策略和药品质量体系,整一个完整的过程强调对产品和生产的认识。QbD能够在一定程度上帮助我们更好地理解产品和工艺,增强单抗制品的稳定性,降低GMP生产的复杂性和成本等。
在细胞培养的基础上,进行抗体表达是抗体药物生产的关键步骤。抗体表达包括转染、筛选和扩增等步骤。
将目标抗体的基因导入到宿主细胞中,实现目标蛋白质的表达。转染类型一般有稳定转染和瞬时转染两种。在单抗细胞株筛选稳定转染前通常通过瞬时转染得到一些抗体,进行质量分析,以初步判断抗体是不是满足需要,或进行一些前期的启动子、密码子等组件的优化。稳定转染中外源DNA既可以整合到宿主染色体中,也可能作为一种游离体存在。在CHO细胞中进行稳定转染时,外源DNA整合到CHO染色体上,未整合的游离态的DNA会随传代而丢失。
常用于哺乳动物细胞转染的方法有磷酸钙法、阳离子脂质体法、阳离子聚合物法和电穿孔法,使用最多的是阳离子脂质体法和电穿孔法。
筛选高表达单克隆细胞株是细胞株构建的关键步骤,衡量标准包括抗体表达量、产品质量、代谢稳定性、细胞稳定性等。当目的基因被转染到宿主细胞内,并按照一定的细胞密度在合适的筛选压力下进行传代培养后便得到了细胞池。细胞池的表达量取决于转染的方式、筛选压力的作用以及目的基因整合情况。由于目的基因整合具有很大的随机性,即使用相同的办法来进行转染和筛选,表达量依然会体现出极大的差异性,接下来就要从细胞池中筛选出高产稳定的细胞株。
CHO细胞转染之后,通常会做一个克隆池(pool),接着进行筛选。筛选策略主要涉及抗生素或药物的代谢途径,常见的筛选试剂包括puromycin、G418、MTX和MSX等。Puromycin为氨基糖甙类抗生素,通过干扰核糖体功能阻断哺乳动物细胞的蛋白质合成,来自链霉菌的pac基因具有解除嘌呤霉素(puromycin)毒性的作用,在筛选的时候,puromycin浓度一般在10~50ug/ml;G418也是一种氨基糖苷类抗生素,是稳定转染最常用的抗性筛选试剂之一,G418在筛选的时候一般浓度范围为200~1000ug/ml;MTX为叶酸拮抗剂,在细胞内经过转换后可抑制DHFR的活性,抑制核酸合成,引起细胞毒性。Schimke等的研究显示,随着MTX浓度的增加,绝大多数细胞死亡,但在极少数幸存下来的抗性细胞中,DHFR基因得以扩增,目的基因拷贝数随之增加,提高了表达量。MTX筛选时,一般浓度范围为25~1000 nmol/L;MSX筛选采用的是谷氨酰胺合成酶基因GS系统压力,谷氨酰胺合成酶(GS)扩增系统是新近发展的更有效的系统,具有更高的扩增效应。MSX筛选时,一般浓度范围为25~500umol/L。
常用的筛选克隆的方法有:有限稀释法(limiting dilution cloning,LDC)、流式细胞仪分选法(fluorescence activated cell sorter,FACS)、半固体培养基筛选法等。其中有限稀释法因为其低成本和易于操作,曾普遍的应用于克隆的筛选。该法是将细胞稀释到极低的细胞密度,并将稀释好的细胞置于96孔板中培养,使孔板中每个孔的理论细胞数小于1个,稀释后使用显微镜对96孔板整板拍照,以确保单克隆。经过一段时间的培养后使用酶联免疫吸附反应选取表达量高的细胞扩大培养。有限稀释法往往需要重复两次,以确保单克隆细胞株的纯度。
另一种开始普遍的使用的方法是流式细胞仪分选法。将带有荧光标记的二抗和分泌抗体的CHO细胞混合孵育,分泌到细胞表面的抗体就能够被流式细胞仪检测到,从而利用其分选功能筛选出分泌多的细胞。为了使分泌到胞外的抗体能够维持在细胞膜表面,还能够正常的使用微囊将细胞以及分泌的抗体包裹起来。目前,又有新的方法运用于流式细胞仪分选,即将绿色荧光蛋白(green fluorescent protein,GFP)的两个基因片段作为报告基因分别构建到抗体重链和轻链的表达载体上进行共转染,当这两个片段都整合到细胞内进行表达时,能够产生绿色荧光蛋白而发出绿色荧光,利用FACS进行分选得到高表达的细胞。使用该方法在转染完成后48h就可以有效的进行检测,可以有效缩短克隆筛选的时间,提高实验效率。
抗体作为临床应用最广泛的治疗性蛋白药物,主要是通过CHO(Chinese Hamster Ovary)细胞培养来表达生产。与前一代的生物技术产品如细胞因子类药物相比,抗体类生物药的主要特征是临床用药剂量大,因此就需要大规模工业化生产来满足市场需求,而CHO细胞培养的工艺优化和规模放大具有较大挑战性。提高细胞培养工艺表达量,扩大细胞培养生产规模,保证表达抗体质量稳定成为目前国内抗体产业界在抗体类蛋白药物规模生产的全部过程中亟待解决的问题。
抗体分离纯化的最大的目的是将抗体与工艺相关杂质和产品相关杂质分离,最终获得高纯度、低潜在危害的抗体药物。
纯化过程中需要去除的工艺相关杂质包括细胞、细胞碎片、宿主细胞蛋白、宿主细胞核酸及培养基与加料液成分,需去除的产品相关杂质最重要的包含抗体片段和聚集体。纯化过程还需具备足够的病毒灭活去除能力,以去除宿主细胞自身表达的内源病毒样颗粒和未及时有效地发现的外源病毒。常用的纯化方法有亲和层析、离子交换层析、凝胶过滤层析等。
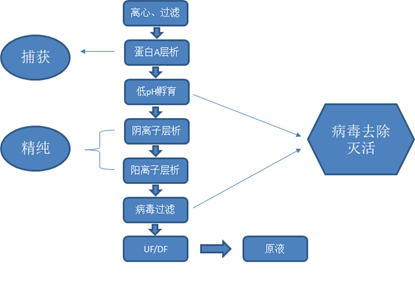
将表达高表达目标抗体的细胞进行破碎,释放出内含的目标抗体。抗体为胞外分泌型表达,纯化的第一步是将培养液上清与细胞及细胞碎片分离开来。在实验室规模,分离可通过简单批次离心完成,但大规模抗体生产主要是采用连续分离的方法,最重要的包含深层过滤、切向流过滤和连续流离心。
深层过滤的优点是简单易用,前期投入低。深层过滤介质的孔径分布较广,内表面积大,可依靠孔径截留和内表面吸附双重作用去除固体颗粒,比单一孔径滤器能处理更大量的固体杂质,且流速更快。
深层过滤系统由一系列的滤器组成,介质的孔径从上游到下游递减(见下图)。深层过滤本身不能达到无菌过滤,所以过滤系统的最后一级常常要一个无菌滤器。深层过滤介质中含有硅藻土,硅藻土在抗体纯化条件下带正电荷,在去除细胞和细胞碎片的情况下,还可以去除部分带负电荷的宿主细胞核酸和宿主细胞蛋白质,起到初步纯化的作用。
深层过滤的缺陷是一次性滤芯使用费高和处理量有限,在处理大体积培养液时需并联多个滤器,费用和占地面积明显地增加,所以深层过滤的试用范围限于100-2000L的中试规模的抗体纯化。

切向流滤器在灌流反应器部分较为常用(见下图),其高切向的流速可减少细胞在膜表面的沉积,从而能够处理大体积细胞培养液而不造成膜的堵塞。
切向流的流速越高,细胞沉积越少,但同时带来的剪切力越高,细胞破碎风险越大。细胞破碎形成的细胞碎片更容易阻塞滤芯,并且细胞破碎释放的胞内蛋白和核酸将大幅度提升后续纯化步骤的压力。
切向流滤器的使用以中空纤维最为普遍,其表面积大,又可以在孔腔内形成高剪切力。中空纤维滤器的工艺放大可通过增加纤维数目完成,简单易行。
在切向流过滤工艺开发和优化方面,需要仔细考虑的包括滤芯化学成分、滤芯表面积、孔径、切向流流速、跨膜压力。
切向流过滤存在死体积,可在过滤后期加人少量缓冲液,降低死体积中的抗体含量,减少抗体损失。切向流过滤处理量大,可用于超过10000L细胞培养的分离,达到每小时近5000L的液体处理量。其局限在于滤器费用较高,过滤时间比较久,并且过滤速度的可控性低。

连续流离心,尤其是采用碟片式离心机进行的连续流离心,是大规模抗体生产中主要是采用的分离模式。
碟片式离心机腔体为锥形,细胞培养收获液在靠近轴部加入,离心后上清液从轴部附近排出,细胞在离心力作用下,在锥形的底部边线附近聚集结块。
腔体定期在细胞聚集处轻微开启,利用腔体内的压力将细胞排出。细胞排出夹带的液体损失可通过离心力(转速)、腔体开启频率和开启时间的优化,控制在5%以下。
连续流离心能够去除绝大部分细胞和细胞碎片。残留的少量细胞碎片可用深层过滤去除。碟片式离心机的优点是体积小,多层碟片形成的沉降面积大,液体处理量大,简单易操作、可靠,使用费低。其缺点是前期设备投资髙,清洗较复杂,并缺少合适的小试模型。
连续流离心机与沉降器一样利用细胞与培养基的密度差实现固液分离。下图显示的是一个商业化的碟片式连续流离心机。细胞培养液自离心机底部进料,在离心力的作用下,细胞在腔体的最外围富集。
上下腔体周期性地短暂打开,释放清除腔体内积累的细胞,这部分细胞返回反应器。细胞培养液上清自离心机顶端收获。连续流离心分离速度快,液体处理量大,可实现高达3600L/d的灌流速度,细胞分离效果高于90%,有很好的应用前景。
连续流离心机的弱点是设备和操作相对复杂,机械和操作故障风险相比来说较高。同时离心时形成的细胞团有可能造成局部营养缺失或副产物积累,其程度和影响需进一步考察。连续流离心机在操作时还需考虑细胞的剪切力耐受性,避免细胞损伤。

亲和层析是利用待分离组分和其特异性配体间具有特异性亲和力,进而达到分离的目的。利用亲和层析等方法将目标抗体与其他杂质分离。常用的亲和层析介质包括蛋白A、蛋白G等。
原理:亲和层析是基于生物分子与其它配基分子之间(如抗原与抗体,酶和底物,激素与受体,核酸中的互补链,多糖与蛋白复合体等)的亲和吸附原理建立起来的。
蛋白质与层析载体上的配体通过共价键、范德华力、疏水力、静电力等作用发生生物学专一性结合形成复合物,共价链接在载体表面的功能团配体上。蛋白质亲和层析技术通过目标蛋白质与配体间通过特异性亲和力吸附而达到分离纯化的目的,具有生物专一性的特点,纯化效率高。
ProteinA是金黄色葡萄球菌的一种膜蛋白,具有与抗体特异性结合的能力,ProteinA亲和层析柱已成为应用广泛的纯化抗体的亲和柱,可从腹水,血清和细胞培养上清或细胞抽提物中分离和纯化多种哺乳动物不同亚型的抗体或包含抗体Fc片段的基因工程重组蛋白。
天然ProteinA由5个IgG结合域和其他未知功能的非Fc结合域组成,分子量42KD,天然ProteinA对IgG的亲和能力很强,可以吸附大量IgG(见下图)。但同时,天然ProteinA的其他非结合域和非目标蛋白结合,这样被洗脱下来的蛋白纯度不够,会影响到后续的试验。
运用基因工程技术,克隆出ProteinA的基因,并对其结构改造,除去了一些不重要的非结合域。偶联这种重组RroteinA的琼脂糖凝胶柱在蛋白纯化中,的确提高了产物的纯度。通常用具备较少B结构域的ProteinA柱能获得高纯度的IgG,洁洗脱条件温和,从而防止蛋白质集聚,保护蛋白活性。
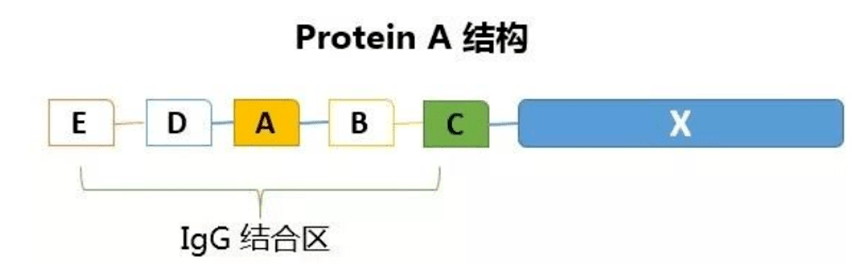
抗体纯化的一个显著特点是ProteinA亲和层析的广泛应用,超过2/3上市抗体品种的生产使用ProteinA进行抗体捕获。ProteinA对抗体重链稳定区Fc有很高的特异性和亲和力,对抗体纯化有很好的通用性。
ProteinA对IgG的高亲和能一步去除培养上清中大部分杂质,包括核酸、宿主细胞蛋白和有几率存在的病毒,抗体纯度达到95%或更高,收率近100%。ProteinA层析操作简单便捷,离心过滤后的细胞培养液可直接加载,不需经过任何处理。
抗体加载后,先用缓冲液淋洗,去除与介质或抗体弱结合的杂质,然后利用强酸性缓冲液(pH3.0-3.5)洗脱ProteinA上结合的抗体,再利用酸性更强的缓冲液(PH约2.0)去除强结合杂质,进行介质再生。ProteinA的清洗能够正常的使用盐酸胍、尿素或低浓度碱液。
ProteinA亲和层析还是一个有效的抗体浓缩步骤,洗脱液中的抗体浓度可达10g/L以上,大大缩小后续精纯的液体处理量。
ProteinA亲和层析工艺开发和优化的重点是抗体载量、停滞时间(流速)、淋洗和洗脱条件。Shukla等考察了14个IgG抗体的ProteinA纯化,发现不同抗体间存在很大的差异,抗体的动态载量相差3倍(10-40g/L),洗脱需要的氢离子浓度相差一个数量级(pH3.0-4.1)。
利用相同的工艺,洗脱峰中宿主细胞蛋白浓度可以从接近完全清除到残留21OOOppm,聚集体浓度可以从1%-20%,说明即使使用类似的平台技术,也需对特定的抗体进行特定的工艺开发和优化。
ProteinA在使用的过程中会发生脱落,脱落的根本原因是细胞培养液上清中含有蛋白水解酶,在上样过程中会对ProteinA进行水解。部分脱落的ProteinA与抗体结合,残留在洗脱液中。由于ProteinA具有免疫原性,需在下游精纯步骤中加以清除。
ProteinA虽然在抗体纯化中被普遍接受,但改进和寻找ProteinA替代物仍是抗体纯化研究的一个重点。
ProteinA在拥有高特异性、高亲和力和高通用性等优点的同时,也具有若干显著缺点,包括费用高、载量低、线性流速有限、需低pH洗脱、ProteinA脱落等。
ProteinA亲和介质比普通纯化介质贵大约10倍,在抗体生产所带来的成本中所占的比重高于其他任何一个原材料。抗体在ProteinA上的载量较低,动态载量通常在20-30g/L,造成介质用量高,进一步提升了使用成本。为控制生产所带来的成本,ProteinA通常会重复使用。
实验证明,利用较温和的清洗步骤,ProteinA介质可以重复使用高达300次。为降低介质成本,大规模抗体纯化一般会用较小的ProteinA层析柱,将单批收获液分成多批进行ProteinA纯化,减小了纯化通量。ProteinA上样是亲和纯化的速度限制步骤。Ghose等建议,可利用双重上样流速缩短上样时间。
在上样初期,当抗体容易到达的结合位点为空时,采用高流速,等到抗体容易到达的位点被占领,抗体一定要通过扩散达到孔内结合位点时,再采用低流速,给抗体更多的扩散时间,达到较高载量。
天然结构ProteinA在强碱清洗时会降解,所有只可以使用低浓度碱液(约0.lmol/LNaOH),尿素或盐酸胍进行清理洗涤,但尿素和盐酸胍废弃液体处理困难,不适于大规模生产,而低浓度碱液在内毒素清除方面的能力有限。
MabSelectSuRe(GE)是一个改构的ProteinA,替换了ProteinA中易在碱性条件下降解(脱酰胺化)的天冬酰胺,从而使得重组ProteinA可以耐受强碱的清洗。
在改进ProteinA的同时,寻找和设计可以替代ProteinA的配体也是一个重要的研究方面。具备替代ProteinA潜力的配体包括多肽、适配子、化学合成配体等,但目前这些配体在结合力、特异性和通用性等方面还不能全方位达到ProteinA水平,离商业化应用尚有距离。在今后的5-10年里,ProteinA还将是占主导位置的抗体捕获介质。
离子交换层析应用最为广泛,利用离子交换层析将目标抗体与其他带电杂质分离。通过调整pH值和盐浓度等条件来实现分离。
特别要注意,蛋白的pI表征的是蛋白表面的净电荷,然而离子交换层析时是蛋白的局部电荷和介质作用,另外离子交换介质吸附的一类物质,如阳离子交换介质吸附带正电荷的物质,所以在纯化过程中,需要配合层析过程进来合理的结合及洗脱过程,才可以做到预期的结果(原理见下图)。

阳离子交换是另一个较常用的抗体捕获方法。至少有4个(Humira、Synagis、Soliris、Zenapax)在欧美上市的治疗性抗体药物采用了这种捕获方法,另外多个处于临床阶段的抗体品种也采用了阳离子交换捕获方法。
阳离子交换与ProteinA相比的最大优势是成本。离子交换介质价格仅为ProteinA的1/10,可重复使用百次以上,并耐受强碱清洗。另外,阳离子交换介质的抗体载量可达100g/L,是ProteinA的2-5倍,可降低介质使用量。
Arunakumari等开发了一个基于阳离子交换抗体捕获、阴离子交换精纯的两步抗体分离方法。在大幅度的降低纯化成本,缩短纯化时间的同时,阳离子交换层析捕获收率达到82%,捕获后抗体纯度超过97%,显示此工艺可成功放大1000倍。
阳离子交换与ProteinA相比有两个劣势,一个是特异性差,另一个是通用性差,需针对不一样抗体和抗体表达工艺来优化,如不同抗体的等电点不同。即使是同一抗体,不同的翻译后修饰也造成不一样的等电点,这些都影响到阳离子交换缓冲液pH的选择。
捕获后的抗体需进一步去除宿主细胞蛋白、宿主细胞核酸、抗体聚集体、抗体片段和脱落ProteinA,以达到对患者安全的纯度。精纯一般都会采用两步层析以保持充足的纯化冗余,但采用一步精纯的工艺也慢慢变得多。
精纯步骤常用的方法有阴离子交换层析、阳离子交换层析、疏水作用层析、羟基磷灰石层析和分子筛,其中阴离子交换和疏水作用采用流穿模式,阳离子交换和羟基憐灰石采用结合-洗脱模式。各个层析步骤的主要杂质去除能力见下表。
两步精纯工艺一般会选择一个流穿模式,一个结合-洗脱模式,而单一步骤精纯一般会选择流穿模式(因为捕获步骤为结合-洗脱模式)。精纯介质一般会用高效介质(直径10-30um)以提髙分离的分辨率,同时提髙抗体的回收率。但采用小粒径介质同时也限制了流速,延长了纯化时间。
离子交换是最常见的精纯步骤。经典的抗体纯化步骤采用流穿的阴离子交换和结合-洗脱的阳离子交换两步精纯。阴离子交换对宿主细胞核酸和宿主细胞蛋白有很好的清除作用,而阳离子交换是去除多聚体和抗体片段的有效步骤。离子交换依靠抗体和杂质表面所带电荷差异将其分离。
抗体的电荷来自多个组成部分。赖氨酸、精氨酸、组氨酸可带负电荷,天冬氨酸、谷氨酸、半胱氨酸可带正电荷,N-端的氨基可带正电荷,C-端的竣基可带负电荷,糖基化上唾液酸可带负电荷。
抗体上的净电荷取决于抗体的等电点和缓冲液的pH,当缓冲液pH低于等电点时,抗体带正电荷;当pH高于等电点时,抗体带负电荷。所以,通过调整缓冲液的pH,能改变其在离子交换介质上的结合和流穿。需要指出的是,虽然一半以上氨基酸本身不带电荷,但它们会通过结构影响抗体的表面电荷,也会影响邻近酸性或碱性氨基酸的电荷。
阴离子和阳离子交换的区分在于介质上的活性基团,阴离子交换介质上的活性基团为阳性(带正电荷),可吸附流动相中的阴性离子,阳离子交换介质上的活性基团为阴性(带负电荷),可吸附流动相中的阳性离子。活性基团还可进一步分为强活性基团和弱活性基团,区分的标准是活性基团上所带电荷是恒定的还是随pH改变的。抗体纯化使用的一般为强活性介质。
在介质上加载相反电荷离子,主要是钠离子或氯离子。平衡液选择的标准是抗体(结合-洗脱模式)或杂质(流穿模式)易于取代平衡时加载的反荷离子吸附在介质上,但吸附又不能太强或不可逆,从而易于洗脱。
样品加载时的缓冲液为低盐溶液,以促进抗体(结合-洗脱模式)或杂质(流穿模式)与反荷离子交换,在介质上的吸附。
对于流穿模式,淋洗可采用平衡液,对于结合-流穿模式,淋洗可采用平衡液或可洗脱弱结合杂质的缓冲液,提高洗脱抗体纯化。
结合-洗脱模式的第三步是洗脱,洗脱依靠的是增加流动相中的盐浓度,增强反荷离子与抗体在介质上的竞争,从而洗脱介质上吸附的抗体和其他物质,洗脱下物质的顺序是从弱结合组分到强结合组分。
通过优化洗脱条件,可增加抗体与杂质在洗脱峰中分离的分辨率,从而有效去除杂蛋白、聚集体、抗体片段等杂质。离子交换的最后一步是再生,利用高盐缓冲液清除结合在介质上的所有物质,并在介质上重新加载反荷离子,准备下一个循环。
离子交换缓冲液中的缓冲体系应该在设定pH范围提供足够的缓冲能力,没有毒性,并且与抗体和介质没有相互作用。通常缓冲组分与介质应带相同电荷,所以不会在介质上吸附。线性洗脱可提高洗脱的分辨率,但在大规模生产中,为保证操作的稳定性,一般采取阶梯洗脱。
越来越多的公司开始使用一步精纯来提高收率和纯化速度,减少相关成本。Kelley等开发了一种一步精纯的弱分离阴离子交换层析(WPC)方法。
弱分离阴离子交换层析操作条件介于流穿与结合-洗脱之间,采用比流穿操作更低的离子浓度,以提高流动相中的杂质与介质的结合能力,提高了本步骤的杂质去除能力。低离子浓度同时也增加了抗体在介质上的吸附,造成部分抗体吸附在介质上(大部分抗体流穿)。
这部分弱吸附上抗体能够最终靠一个短暂的淋洗洗脱,保证抗体收率。Kelley等显示,ProteinA捕获和弱分离阴离子交换组成的两步抗体纯化工艺可满足抗体在宿主细胞蛋白、ProteinA、内毒素和宿主核酸的清除要求。聚集体的清除较为复杂一些。
对于部分抗体品种,聚集体比单体的吸附能力强,聚集体能采用WPC方法去除,但对另外一些抗体品种,单体比聚集体与阴离子交换介质的吸附能力更强,这样品种就只可以通过筛选阴离子交换介质或采用结合-洗脱阴离子交换方式去除聚集体。
抗体纯化技术的另一个进展是膜层析技术日益广泛的应用,尤其是在阴离子膜层析方面。阴离子交换层析膜与填料层析介质使用的是同样的活性基团,但膜层析具有如下几个显著优势。
一是速度快,膜层析通过对流方式,使杂质能够非间接接触到膜上所有的结合位点,开放式模孔结构利用分子扩散,可同时保证高流速和高载量。
二是载量高,膜层析动态载量可高达l-10kg/L膜体积是填料层析的10-100倍。三是体积小,缓冲液用量降低。膜层析回收率可达95%-100%,在宿主细胞蛋白去除、核酸去除及病毒去除能力方面也与填料层析类似,具备替代传统填料层析的优势。膜层析为一次性使用,在成本上比循环使用的填料高。
凝胶层析又称为分子筛层析或凝胶过滤,利用凝胶过滤层析将目标抗体与分子量较大的杂质分离,通过选择正真适合的孔径大小的凝胶来实现分离。凝胶是一种多孔性的不带表面电荷的物质,当带有多种成分的样品溶液在凝胶内运动时,由于它们的分子量不同而表现出速度的快慢,在缓冲液洗脱时,分子量大的物质不能进入凝胶孔内,而在凝胶间几乎是垂直的向下运动,而分子量小的物质则进入凝胶孔内进行“绕道”运行,这样就可以按分子量的大小,先后流出凝胶柱,达到分离的目的。
具有分子筛作用的物质很多,如浮石、琼脂、琼脂糖、聚乙烯醇、聚丙烯酰胺、葡聚糖凝胶等。以葡聚糖凝胶应用最广,商品名是sephadex型号很多,从G10到G200,它的主要应用场景范围是:①分级分离各种抗原与抗体;② 去掉复合物中的小分子物质。如除盐、荧光素和游离的放射性同位素以及水解的蛋白质碎片;③分析血清中的免疫复合物;④分子量的测定。
通过以上步骤,得到初步纯化的目标抗体后,能够直接进行最终纯化。常用的方法有逆流层析、高效液相层析等。
高效液相色谱法(HPLC)可对抗体药物以及重组蛋白药物中的不均一性进行相对有效地分析。比如,尺寸排阻色谱法(SEC)利用了分子空间结构的不同,可对抗体多聚体、片段、PEG化蛋白等样品进行相对有效分析。离子交换色谱(IEC)、疏水色谱(HIC)、反相(RPC)以及亲水色谱法(NPC/HILIC)可对抗体中的带电异构体、结构异构体,或者由于糖基化、脱酰胺化和氧化等作用引起的杂质进行高分辨率分析。
最后一步是将纯化得到的目标抗体制备成抗体药物。这包括药物配方设计、稳定性研究和灭菌等步骤。
根据目标抗体的特性和需求,设计合适的药物配方,包括缓冲剂、保护剂等。抗体药物的经典配方组成为:抗体、用于调节溶液渗透性的赋形剂或是用于冻干粉的冻干保护剂、缓冲液和表面活性剂,其中离子强度调节赋形剂主要由氯化钠和非离子强度调节剂(如蔗糖、海藻糖、甘露醇、麦芽糖和山梨糖醇)组成,冻干保护剂为海藻糖和蔗糖。抗体药物溶液pH范围在4.0-8.0之间,所使用的pH调节剂包括组氨酸、柠檬酸、琥珀酸、醋酸、磷酸盐、谷氨酸、己二酸、天冬氨酸、乳酸、氨丁三醇和2-(N-吗啉)-乙磺酸。表面活性剂主要为聚山梨酯20或聚山梨酯80。
对制备好的抗体药物进行稳定性研究,评估其在不同条件下(如温度、光照等)的稳定性。研究之后发现,某些单克隆抗体药物虽然在体外实验中表现出良好的药物活性,但在进入临床试验阶段却会遭遇到体内活性降低的问题。因此在药物研发的初期就要兼顾其药效动力学的问题。抗体药物的稳定性是影响抗体药效动力学的重要的条件之一,首先抗体的高亲和力与高特异性都需要以稳定的结构为基础,这是其正确行使生物学功能的保障。其次,抗体的稳定性越高,则其新生肽链在细胞内装配时产生错误折叠(mis-folding)的概率越低,可溶性表达量也越高。良好的耐热性所带来的紧凑结构使抗体的蛋白酶切位点更不易暴露,并其影响药品保质期及存放条件,关系到药物成本。目前,提高蛋白质热稳定性的方法主要有非共价修饰、化学修饰、添加蛋白质稳定剂、蛋白质工程,以及在液体状态利用矿化技术直接在蛋白表明产生磷酸钙矿化外壳以提高蛋白质的稳定性。
由此可见,在保证抗体亲和力及表达量等性质不受太大影响的情况下,最大限度上提高其稳定性,对抗体药物的研发具备极其重大的现实意义。
抗体分子自身性质:抗体分子一级结构(蛋白序列)对其聚集性有着重要影响,例如当抗体分子的等电点(pI)过高或过低时决定簇互补区(com‐plementarity-determining region,CDR)都会促进聚集,不同的是较低pI的分子所产生的分子间静电相互作用形成了可溶的聚集物,而较高pI的分子,尤其是当和带有负电荷的容器表面接触时,则会形成沉淀性聚集。例如有研究比较了英夫利昔单抗(infliximab)及其仿制药在强制性降解试验中的表现,发现二者尽管在生产的基本工艺和制作的完整过程上有些许不同,但在该试验中并未表现出明显差异,说明一级结构仍然在很大程度上决定了抗体分子的稳定性。还有研究比较了IgG分子的3 个亚型(IgG1、IgG2、IgG4)在分别经过酸性处理(pH=3. 3)之后的表现,结果IgG1保持了单体形式,而另外两个亚型均表现出二聚化倾向,且当恢复到正常pH时导致IgG4形成聚集体。
温度:高温能够破坏单抗结构,且通常不可逆,进而导致聚集;同时伴随温度增高,脱酰胺和氧化反应发生的概率也增加。当环境和温度下有50%左右的蛋白天然结构展开时,将该温度定义为熔解温度(melting temperature,Tm),每种蛋白都有其Tm,一般该温度范围为40℃~80℃ 之间,而通常生物药品储存运输的温度在2℃~8℃ 之间,远低于该温度。此外过低温也会引起蛋白变性,尤其是在反复冻融的情况下。随着冻融次数增加,会出现缓冲液pH值下降、溶质分子浓缩、水冰界面形成等因素的影响。当0. 5 mg /ml贝伐珠单抗(bevacizumab)溶液经受1~30次冻融循环时,其单体峰通过(size exclusion chromatography,SEC)分析,发现随着循环次数的增加而降低。在类似的研究中,对于固定数量的循环冻融(10个循环),单体峰值随着贝伐珠单抗浓度的增加而降低,这表明在较高浓度下冻融循环的稳定性有所提高。
光照:蛋白质的芳香族残基(以色氨酸残基为主)对光照非常敏感,易发生光氧化形成氧化自由基,随后发生断裂和交联,尤其是紫外光相较于白光影响更大。有研究证实光照对蛋白质二级和三级结构的影响取决于其存在形式,通常冻干粉比液体制剂更能耐受光照。LUIS等通过采用 ICHQ1B(International Council for Harmonisation,ICH Q1Bconditions)推荐值(132万勒克斯小时)和环境光照强度(24万勒克斯小时)评估了两种单克隆抗体的光稳定性,结果显示出巨大的差异,表明单抗药物的光稳定性取决于总的曝光量而不是光强度。SHAH等发现了在ICH光照条件下虽然总体上未观察到明显的抗体结构变化,但CH2结构域任旧存在降解。蛋白质药物主要的光暴露发生在储存运输和给静脉输液患者用药期间,因此在药品应用阶段规避这一因素的影响就显得很重要。
机械应力:在单抗药物生产使用的过程中还会受到机械应力(如搅拌或剪切力)的影响。研究证实由于疏水性基团(主要是巯基)暴露,在该过程中形成了大量规格在1. 5~80 μm不等的聚集体,且聚集水平与气-液界面积呈指数关系。药物制剂在通过输液管或注射器时会产生剪切力作用,已有研究报道了高浓度单抗溶液的剪切稀化现象(剪切力下黏度的降低),这可能与自身聚集的单抗分子解离有关,但目前尚不清楚过滤过程中的剪切力是否会引起其他改变。
浓度:高浓度下的单抗制剂由于黏度增加,分子间相互作用增强也会促进聚集。HAUPTMANN 等发现高浓度仅仅会增加制剂中小颗粒的数量,而分子量大的聚集物反而减少了;NICOUD等则观察到聚集物随浓度增加而增加的现象。有研究指出,虽然降低抗体浓度可以使结合性较弱的聚集体解离,但若不同时改变抗体分子与辅料的比例,则会稀释药物制剂中的辅料(具有保护性的糖类、表面活性剂或精氨酸),影响了溶液本身的pH值和离子强度,导致抗体分子化学稳定性降低。此外,蛋白质分子自身具备聚合倾向,是由分子间的电荷相互作用所主导,且受溶液的离子强度影响。
包装:除对单抗药物固有属性及结构的优化外,也不能忽略在现有生产条件下包装存储方式的影响。蛋白质作为表面活性分子,有吸附到疏水界面的倾向因此导致产品损失、效力降低和潜在的剂量不足。KUMRU等的研究根据结果得出聚氯乙烯(PVC)材质的输液袋(Ⅳ-bags)相较于聚烯烃材质对1 mg / ml的IgG4溶液吸附效果更明显,且颗粒物和浑浊度显著增加,而在加入聚山梨酯20后两种输液袋中的颗粒数降低至接近阴性对照。
造成抗体不稳定性的因素大致划分为化学不稳定性因素和物理不稳定性因素。它们各自对抗体性质产生一定的影响,且彼此相互作用:相关化学反应可导致物理不稳定性,物理不稳定性又可使某些活跃的化学基团产生一些变化,或将有可能相互作用的基团间距离拉近。
氧化作用:化学降解不仅不利于抗体药物的储存,在应用中还会使药物有效性发生损耗。氧化反应(包括二硫键的形成)是最常见的引起化学降解的因素,这种氧化可以发生于氧化剂(例如:光照、过氧化物或者活泼金属)存在的情况下,也可以自发氧化的形式产生。抗体分子中相当一部分氨基酸基团易被氧化,例如甲硫氨酸、组氨酸和半胱氨酸。二硫键的形成即发生在两个半胱氨酸的巯基之间,且类似的氧化不仅可发生在单个分子内部,也可在分子间产生。
脱酰胺作用:另一个引起降解的因素便是脱酰胺化,涉及含天冬酰胺(Asn)和谷氨酰胺(Gln)残基的位点。脱酰胺化类似于酸碱反应,是在临近反应位点的质子供体(通常为丝氨酸或者苏氨酸)参与下形成环酰亚胺中间物,从而破坏抗体本身的结构。例如Asn经脱酰胺化往往生成琥珀酰亚胺,之后很快自发水解为天冬氨酸或异天冬氨酸(Asp)。
水解作用:在抗体分子中另一大化学性质不稳定因素便是二硫键或肽键的断裂。其中在单抗药物正常使用的过程中“单价抗体”的形成就是由二硫键断裂导致。而肽键断裂则形成大量性质和大小不一的小分子量片段,且尚不能确定该现象是由酶解导致。同样的,抗体分子中铰链区的水解机制亦不明确,例如在研究木瓜蛋白酶对单抗裂解的过程中加入蛋白酶抑制剂并不能改变抗体分子的断裂,因此推测可能是由某种非酶解机制导致的该现象。尽管具体机制尚不明确,但抗体分子这一降解途径通常发生在强酸或者高温环境中。
辅料作用:除了上述可能由环境影响发生的降解外,糖类作为常用的辅料亦会对抗体分子产生一定的影响。研究之后发现蛋白质在还原糖作用下可发生糖基化(glycosylation),最终形成酮胺而导致褐变,且从抗体在胞内克隆直至静脉注射,该反应都可能会发生。虽然现在大多厂商以非还原糖作为辅料,但其依然可以降解为还原糖进而影响单抗药物结构和功能。
化学修饰对于单抗药物性质的影响程度取决于其修饰位点。例如,脱酰胺化在Fc段发生所造成的影响很小,但若发生在CDR上则会降低单抗的亲和力及效价;同样的,氧化反应若是发生在Fc段则不仅会降低结合亲和力,更会增加单抗在人体内的清除率以此来降低血药浓度。一些研究表明,化学不稳定性还会导致蛋白构象的改变和分子聚集,例如甲硫氨酸氧化易引起二级结构稳定性改变。
蛋白质变性往往意味着高级结构的展开,上述化学不稳定性改变、外因(温度及pH)等都可能会导致这一结果,并随之降低抗体铰链区的活动性,增加聚集性。聚集性增加是最主要的物理不稳定性改变,聚集体由多个蛋白质分子通过非共价键(范德华力、氢键、疏水和静电相互作用)相链接,而不涉及一级结构和蛋白序列的变化。除此以外还有部分聚集体是通过二硫键等共价键相缔合,称为共价聚集。这两种聚集最终都可能会形成可溶性或不可溶性聚集物。
抗体的聚集大多都是不可逆的,尤其是在后期当形成的聚集物是由大量非天然结构的抗体分子组成的时候。有学者提出了抗体聚集物有可能通过两种途径使正在接受单抗类药物医治的患者产生免疫原性:T细胞依赖途径以及细胞因子激活途径。而且分子量越大的聚集物其免疫原性越强,且糖基化程度、聚集产物来源等亦对其有影响。最终,免疫原性反应不仅大幅度降低了药物的体内疗效,而且易引发Ⅰ型超敏反应。当然,上述现象仅在体外实验及动物模型中观察到,并预测了聚集物在人体内的风险效应,其具体的机制仍然有待探究。
生物技术产品稳定性评估通常包括生物活性分析、分子结构和纯度分析(含降解产物的定量检测)以及相关参数的监测(如外观、pH值等),综合以上数据来对样品的耐热性、聚集性和分子间作用力大小做出评估。在对抗体效价和稳定能力做评估的方法中,除了最常用的间接ELISA法外,还常应用差示扫描量热仪(DSC)测量蛋白质热稳定性,其不但可以得到熔化温度,还能够获得与熔化有关的焓、熵和自由能。逐步发展而来的差示扫描荧光法(DSF)、圆二色(CD)光谱法、动态光散射(DLS)检测技术都朝着高通量、高精度或者对蛋白质水动力学监测等方向改进。此外在蛋白质溶解性预测方面,研究者们相继提出了交叉作用色谱法(CIC)、亲和捕获自身相互作用-纳米粒子光谱法(AC-SINS)或克隆自身相互作用-生物层干涉法(CSI-BLI)等技术,取得了一定的进展,这一些方法评估单克隆抗体在低蛋白浓度下的交叉或自身相互作用的可能性,从而预测单克隆抗体在高浓度下的特性。随着计算机辅助设计在生物大分子开发中的应用,对不确定晶体结构的分子,可通过大量的建模和仿真软件来预测抗体-抗原复合物的三维结构;也可使用不相同的力场进行分子动力学(molec‐ular dynamics,MD)模拟,获得更加多有关结合相互作用、稳定性的详情信息,并使非共价键能(疏水能、静电能、非极性能和结合能)的计算变得更容易。
以上就是抗体药物工艺流程中各个步骤和流程的详细描述。从细胞培养到抗体表达,再到抗体纯化和制剂,每个步骤都是抗体药物生产的全部过程中不可或缺的环节。通过严格的控制和优化每个步骤,能够得到高质量、高纯度的抗体药物,为疾病治疗提供有效的工具。
由生物制品圈&抗体圈主办的“2023生物制品工艺发展大会”将于9月22-23日在上海举办,本次大会将主要聚焦生物制品前沿技术、CMC、生产、工艺等热点话题进行探讨交流,涉及人用疫苗、兽用疫苗、抗体药、细胞治疗、基因治疗等生物制品领域。届时将邀请生物制品领域经验权威的专家进行报告,欢迎各界同仁参会学习交流。
组委会获得报名信息后,根据报名信息进行初筛,并进一步与报名者沟通确认,实现精准邀请,最终有机会进入大会微信群(严格审核通过)。

